
机器学习原子间相互作用势(MLIP)正在改变计算化学与材料科学的格局。MLIP 能够实现原子尺度模拟,将高计算成本的量子化学方法的高保真度与人工智能的高效扩展能力相结合。
然而,在这个交叉领域工作的开发者面临着一个长期存在的挑战:缺乏用于 GPU 加速原子模拟的高效 Pythonic 工具箱。对于需要运行大量同步 GPU 加速模拟等应用场景,当前的软件生态中要么缺少可靠且维护良好的工具,要么依赖多个分散的开源项目。
在过去几年中,用于使用 MLIP 运行原子模拟的可用软件一直以 CPU 为中心。传统上,近邻识别、色散校正、远程相互作用及其相关梯度计算等核心运算仅支持 CPU 计算,而 CPU 计算往往难以满足当代研究对计算速度的要求。在混合工作流程中,尽管模型在 PyTorch 中可通过 GPU 加速,但模拟工具仍基于串行化且依赖 CPU,导致中小型原子系统的高吞吐量模拟很快因 GPU 利用效率低下而成为瓶颈。
虽然多年来开发者一直试图在 PyTorch 中直接实现这些运算,但 PyTorch 的通用设计在应对原子模拟所需的空间与力计算等专门操作时,往往难以充分发挥性能。PyTorch 功能与原子建模需求之间存在的根本性不匹配,引发了一个关键问题:如何才能弥合这一差距?
NVIDIA ALCHEMI(化学和材料创新 AI 实验室)在 Supercomputing 2024 上发布,旨在为化学和材料科学领域的开发者与研究人员提供一套基于 NVIDIA 加速计算平台优化的专用工具集及 NVIDIA NIM 微服务。该工具集包含一系列高性能、批量处理且 GPU 加速的工具,专为在机器学习框架层面实现化学与材料科学研究中的原子模拟而设计。
NVIDIA ALCHEMI 提供涵盖三个集成层面的功能:
- ALCHEMI Toolkit-Ops:一个 GPU 加速的批量常见运算库,专为 AI 赋能的原子模拟任务设计,支持近邻列表构建、DFT-D3 色散校正和远程静电等计算。
- ALCHEMI 工具套件:一套 GPU 加速的模拟基础模组集合,涵盖几何优化器、积分器和高效数据结构,助力实现大规模批量模拟并充分发挥 AI 的优势。
- ALCHEMI NIM 微服务:面向化学与材料科学领域的可扩展、云就绪的专用微服务层,支持在 NVIDIA® 加速平台上进行部署与编排。
本文将介绍 NVIDIA ALCHEMI Toolkit-Ops,这是 ALCHEMI 框架中用于加速批处理的通用操作层。ALCHEMI Toolkit-Ops 利用 NVIDIA Warp 实现加速,并对 AI 驱动的原子建模中的常见操作进行批量处理。这些操作通过模块化且易于使用的 PyTorch API 公开,未来版本还将支持 JAX API,便于与现有的及未来的原子模拟工具实现快速迭代与高效集成。
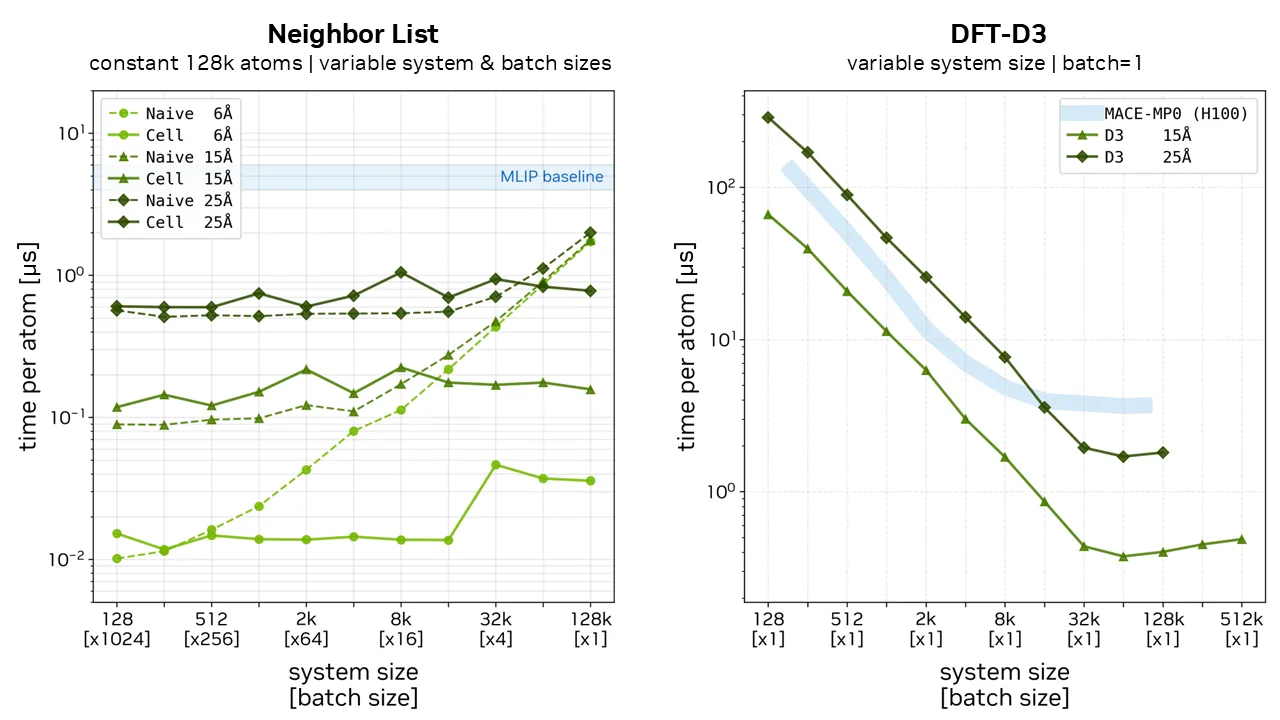
图 1 显示了 ALCHEMI Toolkit-Ops 初始版本中包含的原子模拟加速常用运算。该测试版涵盖了两种近邻列表实现(原生和细胞)、DFT-D3 色散校正,以及远程库伦作用的计算方法(Ewald 和粒子网格 Ewald)。


图 2 展示了 ALCHEMI Toolkit-Ops 中的加速内核与 MACE(cuEquivariance)和 TensorNet(Warp)等主流内核加速模型在实现完全并行性能和可扩展性方面的性能对比。蓝色 MLIP 基准支持与近邻列表和色散校正(DFT-D3)等高级功能进行比较。测试系统包含使用 Packmol 将不同规模的氨分子填充至多种单元中构建的模型。所有实验在 NVIDIA H100 80 GB GPU 上重复运行 20 多次,并对计时结果取平均值。由于 D3 具有长程相互作用特性,DFT-D3 计算未包含 6。

ALCHEMI 工具包 – Ops 生态系统集成
ALCHEMI Toolkit-Ops 旨在与更广泛的基于 PyTorch 的原子模拟生态系统实现无缝集成。我们很高兴地宣布,目前正在与化学和材料科学领域内领先的开源工具推进集成工作:TorchSim、MatGL 和 AIMNet Central。
TorchSim
TorchSim,新一代开源原子模拟引擎,正在采用 ALCHEMI Toolkit-Ops 内核来驱动其 GPU 加速的工作流程。TorchSim 是为 MLIP 时代打造的 PyTorch 原生模拟引擎,能够在单个 GPU 上同时对数千个系统进行批量分子动力学模拟和结构优化。TorchSim 将借助我们优化的近邻列表,实现高吞吐量的批处理操作,同时保持灵活性与高性能。
MatGL
MatGL(材料图库)是一个开源框架,用于为无机、分子和混合材料系统构建基于图的机器学习原子间相互作用势和基础势。通过集成 ALCHEMI Toolkit-Ops,MatGL 能够显著加速基于图的远程相互作用处理,在保持精度的同时实现更快速、更高计算效率的大规模原子模拟。
AIMNet 中心
AIMNet Central 是 AIMNet2 的存储库,该模型是一种通用的机器学习势函数(MLIP),能够高保真地模拟中性、带电、有机及元素有机体系。AIMNet Central 正利用 ALCHEMI Toolkit-Ops,进一步提升其灵活的远程交互模型的性能。借助 NVIDIA 加速的 DFT-D3 和近邻列表内核,AIMNet2 能在不牺牲准确性的前提下,实现对大型周期性系统的更快速原子模拟。
如何开始使用 ALCHEMI Toolkit-Ops
ALCHEMI Toolkit-Ops 入门简单,操作易用。
系统和软件包要求
- Python 3.11 及更高版本
- 操作系统:Linux(主要)、Windows(WSL2)、macOS
- NVIDIA GPU(推荐 A100 或更新型号),CUDA 计算能力 ≥ 8.0
- CUDA 工具套件 12*、NVIDIA 驱动 570.xx.xx*
安装
要安装 ALCHEMI Toolkit-Ops,请使用以下代码片段:
# Install via pip wheel
pip install nvalchemi-toolkit-ops
# Make sure it is importable
python -c "import nvalchemiops; print(nvalchemiops.__version__)"
有关其他安装说明,请参考 ALCHEMI Toolkit-Ops 文档。您可浏览 GitHub 仓库中的示例目录并运行这些示例,以在自有硬件上测试加速效果。
典型的故障排除提示:
- 验证 CUDA 安装及设备可用性:
nvidia-smi、nvcc --version - 确认 Python 版本兼容性:
python --version - 根据需要升级相关依赖项:
pip list | grep torch和pip list | grep warp
功能亮点
本节深入探讨 ALCHEMI 工具包的三个初始特性:高性能近邻列表、DFT-D3 色散校正以及远程静电相互作用。
近邻列表
近邻列表构建是原子模拟的支柱,支持利用局部或半局部 MLIP 计算能量和作用力。ALCHEMI Toolkit-Ops 在 PyTorch 中可实现优异的 GPU 性能,为许多中小型原子系统或单个大型原子系统批量处理时,达到每秒数百万原子的性能扩展。
功能
- O (N) (单元列表) 和 O (N*) (朴素) 算法均采用批量处理方式,
- 为具有任意晶胞维度和部分周期性的三斜晶胞提供周期性边界支持,
- 支持端到端的计算图编译,
- 并与 PyTorch 实现直接的 API 兼容。
API 示例
import torch
from nvalchemiops.neighborlist import neighbor_list
# Water molecule
water_positions = torch.tensor([
[0.0, 0.0, 0.0], # O
[0.96, 0.0, 0.0], # H
[-0.24, 0.93, 0.0], # H
], device="cuda", dtype=torch.float32)
# Ammonia molecule (NH3)
ammonia_positions = torch.tensor([
[0.0, 0.0, 0.0], # N
[1.01, 0.0, 0.0], # H
[-0.34, 0.95, 0.0], # H
[-0.34, -0.48, 0.82], # H
], device="cuda", dtype=torch.float32)
# Concatenate positions for batch processing
positions = torch.cat([water_positions, ammonia_positions], dim=0)
# Create batch indices (0 for water, 1 for ammonia)
batch_idx = torch.cat([
torch.zeros(3, dtype=torch.int32, device="cuda"), # Water
torch.ones(4, dtype=torch.int32, device="cuda"), # Ammonia
])
# Define cells for each molecule (large enough to contain them without PBC)
cells = torch.stack([
torch.eye(3, device="cuda") * 10.0, # Water cell
torch.eye(3, device="cuda") * 10.0, # Ammonia cell
])
# non-periodic molecule case
pbc = torch.tensor([
[False, False, False], # Water
[False, False, False], # Ammonia
], device="cuda")
# Cutoff distance in Angstroms
cutoff = 4.0
# Compute neighbor list; here we explicitly request a batched cell list algorithm
neighbor_matrix, num_neighbors, shift_matrix = neighbor_list(
positions, cutoff, cell=cells, pbc=pbc, batch_idx=batch_idx, method="batch_cell_list"
)
print(f"Neighbor matrix: {neighbor_matrix.cpu()}") # [7, num_neighbors.max()]
print(f"Neighbors per atom: {num_neighbors.cpu()}") # [7,]
print(f"Periodic shifts: {shift_matrix.cpu()}")
DFT-D3 色散校正
逼真的分子建模必须充分考虑范德华相互作用,而标准的DFT泛函并未系统地描述这种作用。DFT-D3通过引入经验性的成对校正,能够显著改善常见DFT泛函在结合能、晶格结构、构象分析和吸附研究方面的表现。
功能
- Becke-Johnson (BJ) 有理阻尼变体
- 支持批量与周期性计算
- 支持截断距离的平滑处理
- 支持联合的能量、力与维里计算
API 示例
from nvalchemiops.interactions.dispersion import dftd3
batch_ptr = torch.tensor([0, 3, 7], dtype=torch.int32, device="cuda")
atomic_numbers = torch.tensor(
[6, 1, 1, 7, 1, 1, 1], dtype=torch.int32, device="cuda"
)
# For this snippet, assume d3_params is loaded as:
# d3_params = D3Parameters(rcov=..., r4r2=..., c6ab=..., cn_ref=...)
# Users can refer to the documentation to source DFT-D3 parameters
# and understand the expected data structure
d3_params = ...
# call the DFT-D3 functional interface
energy, forces, coordination_numbers = dftd3(
positions=positions,
numbers=atomic_numbers,
a1=0.3981, a2=4.4211, s8=0.7875, # PBE parameters
neighbor_matrix=neighbor_matrix,
neighbor_matrix_shifts=shift_matrix,
batch_idx=batch_idx,
d3_params=d3_params
)
print(f"Energies: {energy.cpu()}") # [2,]
print(f"Forces: {forces.cpu()}") # [7, 3]
限制
当前实现仅计算了两个主体项(C6 和 C8),未包含三个主体的 Axilrod-Teller-Muto(ATM/C9)贡献,这通常会导致色散能被高估。
远距离静电相互作用
精确的静电相互作用建模对于涉及离子/带电物种和极性系统的模拟至关重要。目前,MLIP 通常采用在短程模型中学习库伦相互作用的方法。若系统低估远距离库伦效应,将导致结合能、溶解结构和界面现象的准确性下降。
ALCHEMI Toolkit-Ops 提供完全 GPU 加速的 Ewald 求和方法(包含标准 Ewald 和粒子网格 Ewald(PME)),可在 PyTorch 中实现 GPU 加速、高效且精确的长程静电相互作用计算。
对于大型周期系统,基于 Ewald 的方法将静电相互作用分解为短程和长程两部分,每部分在最适合计算效率的域中进行求解。ALCHEMI 工具套件 – Ops 提供双截断策略,相较于原生的全对方法,可显著降低冗余的邻近查询和内存开销,从而在现代 GPU 上实现带电系统的高吞吐量模拟。用户可根据具体性能与精度需求,在适用于小型系统的标准 Ewald 方法与适用于大型周期系统的 PME 方法之间灵活选择。
功能
- Ewald 求和法
- 采用 B-样条的粒子网格 Ewald (PME)
- 支持批量处理与周期性系统
- GPU 优化计算,结合 cuFFT 实现高效的倒易空间评估
- 集成 PyTorch,为端到端可微分工作流提供原生张量支持
API 示例
from nvalchemiops.interactions.electrostatics import particle_mesh_ewald
# charges for each atom are randomly generated here
atomic_charges = torch.randn(
positions.size(0), dtype=torch.float32, device="cuda"
)
# compute energy and forces with particle mesh ewald
energy, forces = particle_mesh_ewald(
positions,
atomic_charges,
cells,
alpha=0.3, # adjust Ewald splitting parameter
batch_idx=batch_idx,
neighbor_matrix=neighbor_matrix,
neighbor_matrix_shifts=shift_matrix,
compute_forces=True
)
print(f"Energy: {energy.cpu()}") # [2]
print(f"Forces: {forces.cpu()}") # [7, 3]
深入了解 ALCHEMI Toolkit-Ops
ALCHEMI Toolkit-Ops 借助 NVIDIA GPU 上可访问的高性能原子建模工具,为科研社区提供支持。如需加速化学与材料科学模拟,可访问 NVIDIA/nvalchemi-toolkit-ops GitHub 代码库,查阅 NVIDIA ALCHEMI Toolkit-Ops 文档,或探索 示例图库。当前测试版 ALCHEMI 工具套件 – Ops 聚焦于高效近邻列表、色散校正及远程静电计算。未来版本将陆续推出新功能与性能优化,敬请期待。
致谢
我们要感谢Shyue Ping Ong教授、Olexandr Isayev教授;以及TorchSim委员会成员Abhijeet Gangan、Orion Archer Cohen、Will Engler和Ben Blaiszik与我们合作将NVIDIA ALCHEMI Toolkit-Ops引入他们的开源项目。同时,也感谢NVIDIA的Wen Jie Ong、Piero Altoe和Kibibi Moseley在准备本文过程中提供的帮助。




