
基础模型 正在重塑计算生物学。预训练在大量蛋白质或基因组序列的语料库上的模型,如 ESM2 (一种蛋白质语言模型)和 Evo 2 (一种 DNA 语言模型),捕获生物序列的统计规律。它们可以很好地转移到各种下游任务,包括结构预测、变异效应和功能注释。
然而,让这些模型适应特定任务并非易事:在数十亿个参数下,无论是在优化器状态和检查点的计算和存储中,完全微调很快都变得不切实际。
低秩自适应 (LoRA) 直接解决了这一挑战。通过冻结预训练的主干并仅训练一小部分低秩适配器矩阵,LoRA 可以在训练约 1% 的参数时,在单个工作站 GPU 上拟合单个十亿级规模的模型及其适配器状态,从而在许多任务中达到完整的微调质量。
为降低构建这些工作流程的难度,NVIDIA BioNeMo Recipes 提供了基于熟悉的 PyTorch、Hugging Face 和 Megatron-Bridge 模式构建的分步训练方法。NVIDIA Transformer 引擎 (TE) 和横向扩展策略等以性能为导向的组件在有回报的地方进行集成,但配方本身仍保持可读性。
本文将介绍两个案例研究,其中展示了相同的参数高效配方如何在单个 NVIDIA RTX 6000 Blackwell 工作站版 GPU 上应用于各种生物模态:
- 用于蛋白质二级结构预测 (PSSP) 的 ESM2-3B plus LoRA
- 用于 DNA 剪接位点分类的 Evo2-1B 和 LoRA
NVIDIA BioNeMo Recipes 中提供了用于自定义或重现这些结果的所有源代码。
LoRA 如何实现大规模微调
在深入探讨案例研究之前,请快速回顾一下该方法。完全微调需要大量资源,因为它需要存储和更新所有模型参数及其优化器状态,而随着模型的扩展,这很快就变得不切实际。
LoRA 是一种实用的方法,可以微调大型预训练 Transformer,而无需为所有模型参数更新或存储优化器状态。LoRA 背后的核心理念是,LoRA 不是更新稠密模型的权重矩阵 \(W\),而是以并行方式添加新的可训练低秩矩阵 \(W=BA\),并使 \(W\) 保持冻结。这大大减少了可训练参数的数量和优化器/ 内存占用。
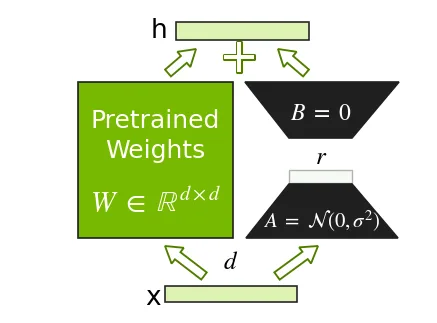
LoRA 由一组权衡容量、稳定性和成本的超参数进行参数化。秩 \(r\) 控制添加的低秩矩阵的大小,进而控制可训练参数的数量。目标模块会指定哪些层可接收适配器,并提供注意力和 MLP 投影等常见选择。对于小型数据集,可以启用 LoRA dropout 作为正则化的另一种形式。
虽然以下两个案例研究在模态 (蛋白质与 DNA) 、任务类型 (标记分类与序列分类) 和底层架构 (转换器与条纹狗) 方面各不相同,但它们都使用相同的 LoRA 配方模式。
用于蛋白质二级结构预测的 ESM2-3B
PSSP任务是为蛋白质序列中的每个氨基酸分配结构标记。二级结构标签可描述局部主干构象(螺旋和链)而无需进行完整的3D结构预测。对于许多蛋白质而言,这些局部模式与功能基序和全局折叠组织相关。
PSSP 是生物学中许多下游应用的核心构建块。由于局部结构与蛋白质功能密切相关,PSSP 可以提供有用的功能上下文。此外,这些预测还可以为三级结构预测、溶剂可及性预测、蛋白质相互作用预测以及与结构类或领域相关的预测提供信息。
在建模层面,PSSP 是一个标记分类问题:输入是氨基酸序列,输出是每个残基的结构标签。
有两种常见的评估变体,仅在标签空间方面有所不同:
- Q3 (3-state):H (Helix) 、E (Strand/Sheet) 、C (Coil/Loop)
- Q8 ( 8 状态) :H (α-helix) 、B (β-bridge) 、E (β-strand) 、G (310 helix) 、I (π-helix) 、T (turn) 、S (bend) 、C (coil/other)
ESM2-3B 是一个 30 亿参数的蛋白质语言模型,因此完全微调通常需要大量计算和内存。LoRA 仅通过训练少量额外参数使适应变得切实可行,同时仍然在 PSSP 上实现强大的性能。
BioNeMo 方法中的 ESM2 和 PEFT ( TE 加速)
该团队通过添加轻量级每残差分类头 (适用于 Q3/ Q8 标签) 并通过 PEFT 库训练 LoRA 适配器,同时冻结预训练的主干权重,从而针对 PSSP 微调了 ESM2-3B。对于数据,我们使用了由“传送门 6 ( Porter 6)”模型的作者发布的精选拆分,并在其提供的测试集上报告了结果。为了更大限度地提高吞吐量,我们启用了 TE 和序列打包,并在一个 NVIDIA RTX 6000 Blackwell 工作站版 GPU 上运行了完整的训练工作流,只需不到一小时。
以下片段改编自 BioNeMo Recipes ESM2+ PEFT 示例,展示了如何加载与 TE 兼容的 ESM2 模型,并将 LoRA 适配器连接到融合查询/ 键/ 值 (QKV) 预测:
import peft
import torch
from transformers import AutoConfig, AutoModelForTokenClassification
# Load config and token-classification model (use a local checkpoint path or HF model ID, e.g. nvidia/esm2_t36_3B_UR50D).
config = AutoConfig.from_pretrained("nvidia/esm2_t36_3B_UR50D", trust_remote_code=True)
model = AutoModelForTokenClassification.from_pretrained(
"nvidia/esm2_t36_3B_UR50D", config=config, trust_remote_code=True, dtype="bfloat16"
)
peft_config = peft.LoraConfig(
task_type=peft.TaskType.TOKEN_CLS,
inference_mode=False,
r=8,
lora_alpha=16,
target_modules=["layernorm_qkv"],
bias="none",
)
peft_model = peft.get_peft_model(model, peft_config)
peft_model.to("cuda", dtype=torch.bfloat16)
然后,您可以将此 PEFT 模型插入训练循环。完整的 recipe 包括 dataloader、loss 和 optimizer 设置。
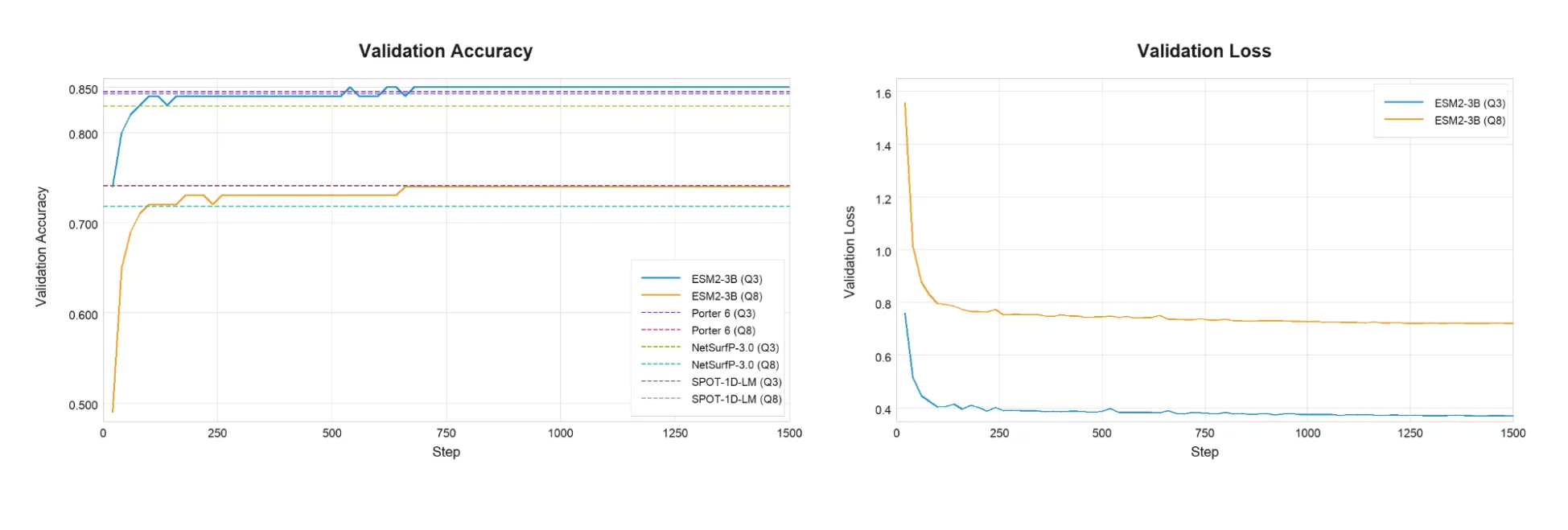
表 1 总结了 ESM2-3B+ LoRA 模型的 Q3/ Q8 测试准确性,以及在《 Porter 6》论文中报告的已发布的基准。表 1 报告了 ESM2-3B 前五个验证检查点的平均分。
| 模型 | Q3 准确率 (%) | Q8 精度 (%) |
| ESM-2 3B 加 LoRA (前五项验证平均值) | 84.80 | 74.30 |
| Porter 6) | 84.56 | 74.18 |
| NetSurfP-3.0 | 82.92 | 71.84 |
| SPOT-1D-LM | 84.30 | 74.09 |
总体而言,LoRA 微调的准确性可与其他先进的 PSSP 方法相美。图 2 显示了验证损失和准确性与微调步骤的对比情况。
序列打包如何提高利用率和吞吐量?
蛋白质数据集通常包含不同长度的序列。如果简单地对其进行批量处理 (填充的 BSHD 格式) ,则会将其填充到批量中的最大长度,并且大部分令牌会变成填充。这会浪费注意力层和 MLP 层内部的计算和内存带宽。
序列打包 (packed/flattened THD 格式) 通过仅连接非填充令牌并使用累积长度元数据跟踪每个序列边界来减少这种浪费。因此,注意力/ MLP 核函数在真实 token (而非填充 token) 上运行。如需更深入地了解打包在实践中的工作原理 (及其与 TE 打包格式的交互方式) ,请参阅使用 PyTorch 和 NVIDIA BioNeMo Recipes 扩展生物学 Transformer 模型。图 3 显示了使用 THD 与 BSHD 进行微调时的吞吐量 (令牌/ 秒) 。
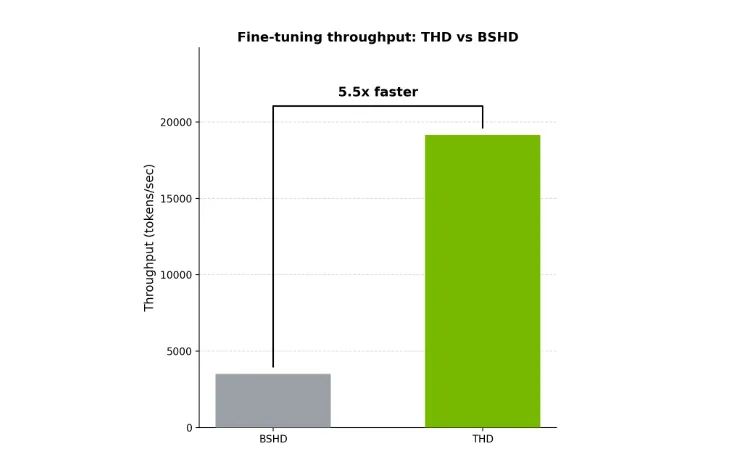
在此设置中,从 BSHD 切换到 THD 可将 token/ 秒提高约 5.5 倍,主要原因是消除了填充开销。实现的加速在很大程度上取决于序列长度分布、微批量大小和 GPU。
除了吞吐量,THD 打包还提高了内存效率。它减少了在填充令牌上花费的激活或注意力工作量,因此 GPU 显存流量和计算的更大部分都流向有用 (非填充) 令牌。
对于相同的输入序列和批量大小,THD 通常比 BSHD 占用更少的内存,因为它避免了实现填充令牌。在实践中,节省的空间用于增加每个步骤处理的真实令牌数量。
用于 DNA 剪接位点分类的 Evo2-1B
Evo 2 是一个生成式 DNA 基础模型,在涵盖所有生命领域的基因组序列上进行训练。在架构上,它建立在条纹 Hyena 块上,混合了状态空间式的长卷积算子和较少数量的注意力层。这使其能够高效处理长 DNA 上下文。就像 ESM2 从氨基酸序列中学习蛋白质“语法”一样,Evo2 直接从核酸序列中学习基因组规律,这些序列会转移到各种下游任务:变异效应预测、调控元素分类和 (此处关注的) 剪接位点识别。
什么是接头位点分类?
剪接是细胞过程,可从 mRNA 前分离出内含子,并将外显子连接在一起。边界由两个短序列基序定义:内含子 5 ° 端的供体站点 (内含子开始,通常为 GT) 和 3 ° 端的受体站点 (内含子结束,通常为 AG) 。
从原始 DNA 中识别这些位点比仅匹配二核酸基序困难得多。相同的 GT/ AG 模式在整个基因组中出现,只有一小部分是功能性拼接位点。有用的预测器必须学习候选位置周围的更远距离上下文。
我们使用了 Nucleotide Transformer 下游任务数据集中的 splice_sites_all 任务。每个示例都是一个固定长度的 600 bp DNA 窗口,标签是描述中心位置 (无拼接、受体或供体) 的三个类别之一。该基准测试提供了约 3 万个训练/ 约 3,000 个测试示例,并且大致实现了类别平衡。
在建模层面,这是一个序列分类问题:每个输入序列只有一个标签,与 PSSP 中的每个 token 标签形成对比。
BioNeMo 食谱中的 Evo2+ LoRA
该团队通过对 Megatron Hyena 模型进行子分类,在均值池化隐藏状态之上添加一个小的序列分类头,从而对 Evo2-1B 进行了微调,以实现拼接点分类。然后,在主干注意力、MLP 和 Hyena-mixer 投影方面对 LoRA 适配器进行训练。预训练的主干权重保持不变;只有 LoRA 适配器和分类头接受训练。
为了将 LoRA 的贡献放在上下文中,我们使用相同的数据训练了两种配置并进行了比较:
- 只有头部的基准:主干冻结;没有适配器,只有分类头部是可训练的。可训练参数总数:约 370 万 (模型的 0.33%)
- LoRA+ 头: 冻结的主干;列出的目标模块上的 LoRA 适配器,分类头可训练。可训练参数总数:约 1600 万 (占模型的 1.42%)
表 2 显示了对举行的 3K 示例的测试准确性。
| 模式 | 可训练参数 | 可训练分数 | 测试准确性 |
| Head only | 3,697,923 | 0.33 % | 52.3 % |
| LoRA plus Head | 15,985,923 | 1.42 % | 96.6 % |
差距很大:只有约 1% 的参数可训练,LoRA 几乎可以恢复预训练 Evo2 主干所持有的关于拼接的所有信号,而仅依靠池化还远远不够。LoRA 模型的大部分残差误差都在供体 – 受体方向上。这是可以预期的,因为这两种基序都具有 GT/ AG 的双核酸结构,并且需要更广泛的上下文来消除歧义。
整个工作流在单个 RTX 6000 工作站版本上端到端运行,大约需要一小时。
以下代码段从风格上反映了 ESM2 示例:它加载 Evo2 主干,通过 Hyena 模型子类连接分类头,并在注意力、MLP 和 Hyena-mixer 投影上配置 LoRA 适配器。
from bionemo.evo2.models.evo2_lora import Evo2LoRA
from evo2_classifier import (
Hyena1bClassifierProvider,
HyenaForSequenceClassification,
)
# Backbone provider: a HyenaModel subclass with a small classification head
# (LayerNorm → Linear → GELU → Dropout → Linear) on top of mean-pooled hidden states.
model_provider = Hyena1bClassifierProvider(
num_classes=3, # no-splice / acceptor / donor
classifier_dropout=0.1,
pool="mean",
)
# LoRA adapters on attention (linear_qkv, linear_proj), MLP (linear_fc1, linear_fc2),
# and the Hyena mixer (dense_projection, dense). The classification head is kept
# trainable via the skip_freeze_modules pattern; everything else is frozen.
peft = Evo2LoRA(
target_modules=[
"linear_qkv", "linear_proj",
"linear_fc1", "linear_fc2",
"dense_projection", "dense",
],
dim=16,
alpha=32,
dropout=0.1,
skip_freeze_modules=["*classification_head*"],
)
Megatron-Bridge 预训练入口点可处理分布式训练、优化器、调度器、检查点、数据加载和日志记录。
要端到端启动微调运行,recipe 会公开一个 CLI:
torchrun --nproc_per_node=1 evo2_classifier.py \
--train-jsonl splice_train.jsonl \
--val-jsonl splice_val.jsonl \
--test-jsonl splice_test.jsonl \
--base-ckpt-dir evo2_1b_bf16_mbridge \
--result-dir splice_run \
--experiment-name lora_finetune \
--num-classes 3 \
--seq-length-tokens 600 \
--train-iters 1000 \
--global-batch-size 32 --micro-batch-size 32 \
--lr 5e-4 --min-lr 5e-5 --warmup-iters 30 \
--lora-finetune --lora-dim 16 --lora-alpha 32 --lora-dropout 0.1
交换 --lora-finetune 并增加批量大小,以重现仅头部的基准。数据、优化器、调度器和评估保持不变。
有关完整的训练循环、数据集代码、参数计算和评估实用程序,请参阅 Evo2 LoRA 微调 Notebook。
开始微调生物基础模型
在两种截然不同的生物模态 (蛋白质 ESM2 和 DNA Evo2) 中,可以使用相同的参数高效配方。您可以在单个工作站 GPU 上冻结预训练的主干,训练一个小型 LoRA 适配器和一个任务特定的头,并恢复可与完全微调或专用模型相美的准确性。
对于 ESM2-3B,LoRA 将 PSSP 性能引入与强大的已发布基准 (如 Porter 6 和 SPOT-1D-LM) 相同的范围,而 TE 和 THD 序列打包使在单个 NVIDIA RTX 6000 Blackwell 工作站版 GPU 上进行训练切实可行。对于 Evo2-1B,相同的方法将拼接位点分类从 ~52% 的固定骨干基准提升到 ~97% 的测试准确率,同时仅训练约 1.4% 的参数。
如果周围的训练堆栈 ( TE、Megatron-Bridge、Packed sequences、PEFT) 能够很好地集成,包含数十亿个参数的生物基础模型现在可以在普通硬件上进行调整。NVIDIA BioNeMo 方法 旨在将该集成作为默认集成,而非例外。
要开始使用 LoRA、TE 和可扩展的 PyTorch 工作流微调生物基础模型,请查看 NVIDIA BioNeMo Recipes。