
生物分子结构预测以及与 OpenFold3 等模型的联合折叠现已成为主流的大规模工作负载,为药物研发和蛋白质设计提供支持。它们越来越多地由 AI 智能体提供端到端驱动。要让智能体良好运行该工作流,每个步骤都需要快速且可扩展:多序列比对 (MSA) 生成、联合折叠推理、服务和多 GPU 横向扩展。任何地方的瓶颈都会限制整体吞吐量。
速度和内存效率对于关键的药物研发工作流程 (例如虚拟筛选和大分子组合预测) 至关重要。在虚拟筛选中,针对一个或几个蛋白质标筛选数百万到数十亿种化合物。虽然联合折叠模型通常会给出最佳预测结构,但运行成本可能很高,因此无法用于虚拟筛选应用。这正是 NVIDIA 加速的关键所在,使得 OpenFold3 和相关方法能够在大型化合物库中大规模部署成为可能。
在预测涉及多种蛋白质和数千个氨基酸残基的大型分子组合时,速度也很重要,因为联合折叠模型的运行时随残基数量呈立方体扩展。然而,更大的挑战是显存使用,因为单 GPU 显存可能会受到限制,从而对单次预测的复合体大小施加硬性限制。降低内存需求以及在多个 GPU 之间分配预测任务的方法将启用目前根本不可行的定性新应用程序。
NVIDIA 已经构建了一些工具,用于加速和提高结构预测和联合折叠工作流程每个步骤的效率。借助 NVIDIA BioNeMo Agent Toolkit,智能体可以无缝访问加速生物学和化学工作流所需的工具。在本文中,我们将详细介绍 NVIDIA B300 和 H100 GPU 上每个阶段的加速情况,然后展示如何通过智能体执行这些阶段 (请参见下图 1) 。
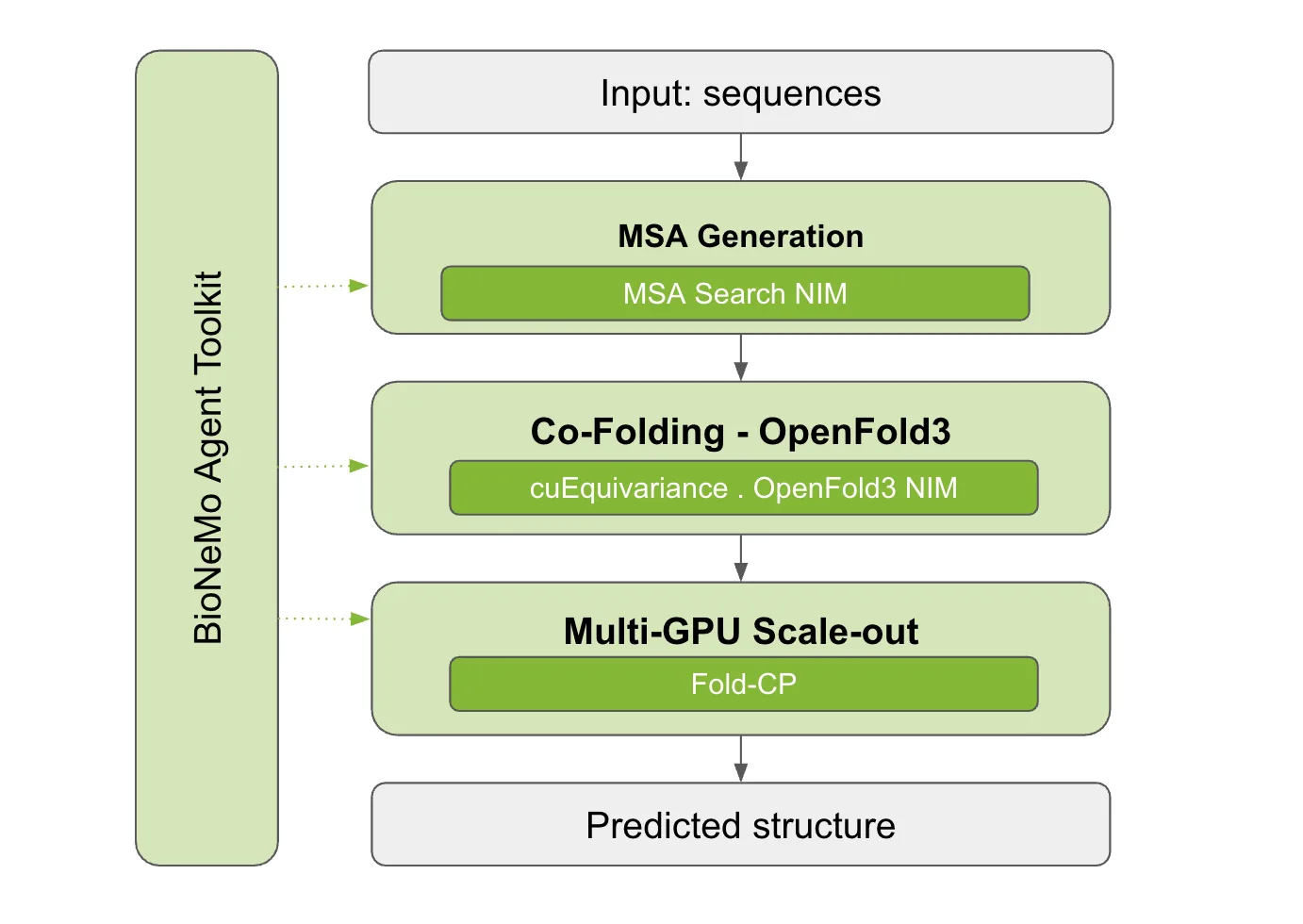 图 1. NVIDIA 加速的结构预测工作流:GPU MSA、cuEquivariance、优化推理和 Fold-CP,全部由 BioNeMo Agent Toolkit 编排
图 1. NVIDIA 加速的结构预测工作流:GPU MSA、cuEquivariance、优化推理和 Fold-CP,全部由 BioNeMo Agent Toolkit 编排使用 GPU MSA 消除 MSA 瓶颈
对于联合折叠模型而言,构建 MSA 传统上是受 CPU 限制的步骤,可能会控制挂钟时间。MMseqs2-GPU 可将同源搜索转移到 NVIDIA GPU 上,从而减少此瓶颈,同时在 NVIDIA Hopper 和 NVIDIA Blackwell 架构上根据序列长度进行扩展。
最新的 GPU 加速版本增加了专门针对 Hopper 和 Blackwell 的优化,包括对 NVIDIA Grace 系统上大于 GPU 内存的数据库搜索的高效支持,以及CUDA 13.2中提供的改进的 Blackwell DPX 指令的额外加速。这些 GPU 贡献已上传到主MMseqs2 资源库,以便整个社区都可以从加速中受益。
MSA Search NIM 使用 MMseqs2-GPU,其 Nature Methods 论文 报告称,在单个 L40S 上,比 CPU JackHMMER 的对齐速度快 177%。在我们的基准测试中,该阶段可以平滑地扩展到 H100 和 B300 GPU 上的 1 万个 token (请参见下面的图 2) 。MSA Search NIM 可以直接调用、自行托管,也可以在智能体工作流中作为工具打包。
# Add the MSA Search NIM skill to your agent (browse all: add --list)
npx skills add NVIDIA-BioNeMo/bionemo-agent-toolkit --skill msa-search-nim --agent claude-code
# Use hosted API on build.nvidia.com (nothing to download)
# You can also use the skill to download the NIM container and provide self-hosted API endpoint. We don't cover that in this tutorial.
export NVIDIA_API_KEY=<key from build.nvidia.com>
# Just prompt the agent:
# You have to download a sample target.fasta file. You can prompt the agent to download it for you or point to an already existing file.
"Build an MSA for the sequence in target.fasta with the MSA Search NIM."
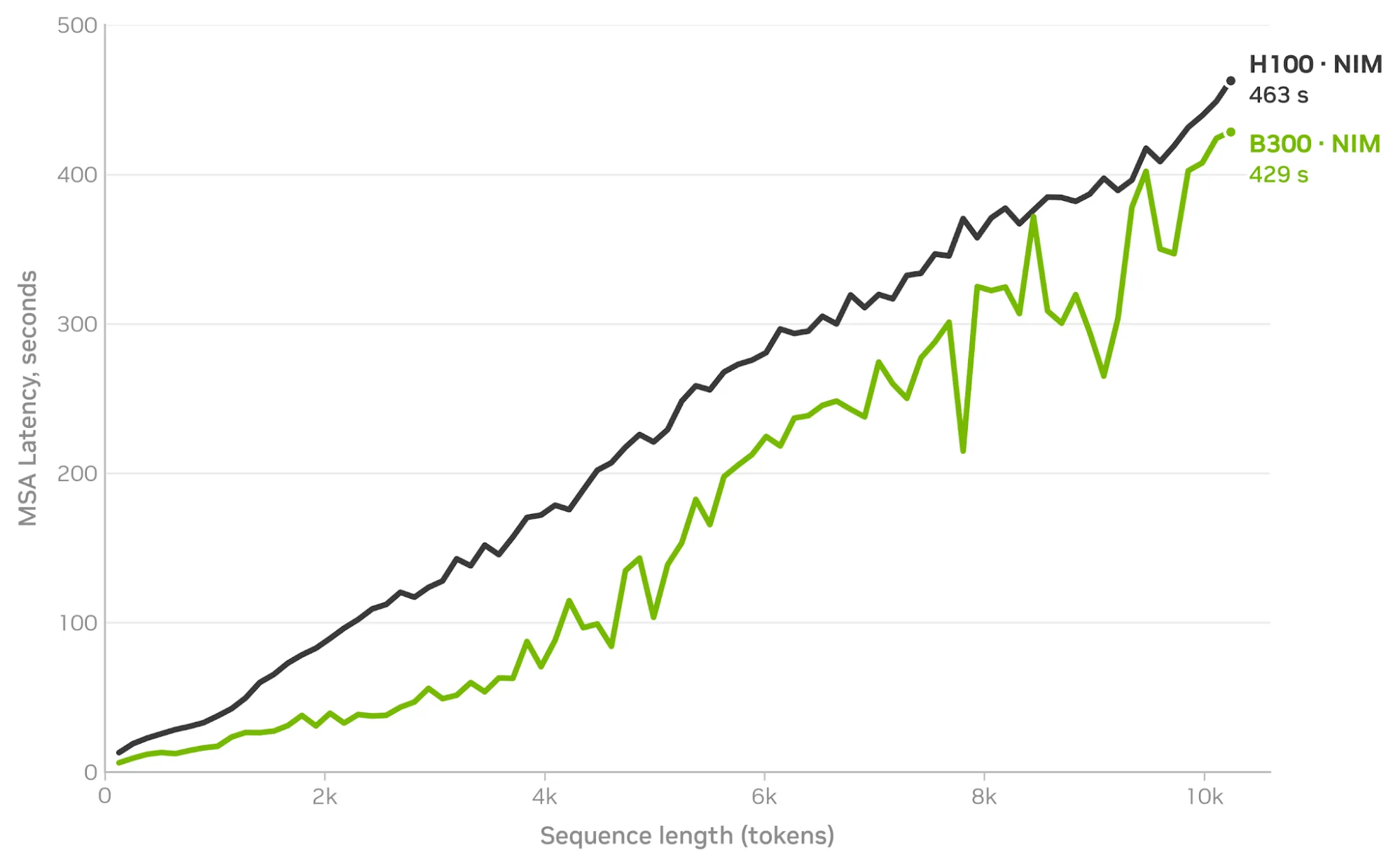 图 2. 在 H100 和 B300 上,MSA 阶段延迟几乎呈线性扩展,达到 1 万个以上的 token (约 1 万个 token 时,延迟为 429 秒,对比为 463 秒)
图 2. 在 H100 和 B300 上,MSA 阶段延迟几乎呈线性扩展,达到 1 万个以上的 token (约 1 万个 token 时,延迟为 429 秒,对比为 463 秒)使用 cuEquivariance 和 OpenFold3 NIM 以 SOTA 速度折叠
cuEquivariance 是一个用于原子建模的 CUDA-X 几何学习基元库,可提供主导联合折叠的三角形注意力、三角形乘法和注意力对偏差内核的加速版本。在 B300 上,它可将延迟最多降低 ~3× (请参阅下表 1)。
| 序列长度 | PyTorch ( OSS) | cuEquivariance | 加速 |
| 1024 ( H100) | 79.4 秒 | 41.6 秒 | 1.9% |
| 1024 ( B300) | 49.1 秒 | 27.3 秒 | 1.8% |
| 1536 ( B300) | 149.0 秒 | 56.2 秒 | 2.7% |
| 2048 ( B300) | 300.1 秒 | 97.6 秒 | 3.1% |
cuEquivariance 内核直接集成到 OpenFold3 (作为可选依赖项提供) 、OpenFold2、RosettaFold3、Protenix 和 Boltz 等 OSS 模型中。
由于这些加速上传到了这些 OSS 模型中,研究人员只需在 NVIDIA GPU 上运行他们已经使用的模型,就能自动获得加速。CuEquivariance 内核还将最大序列长度扩展到约 5.9 k 个令牌,而 PyTorch 的显存不足超过约 1.5 k – 2.5 k 个令牌。
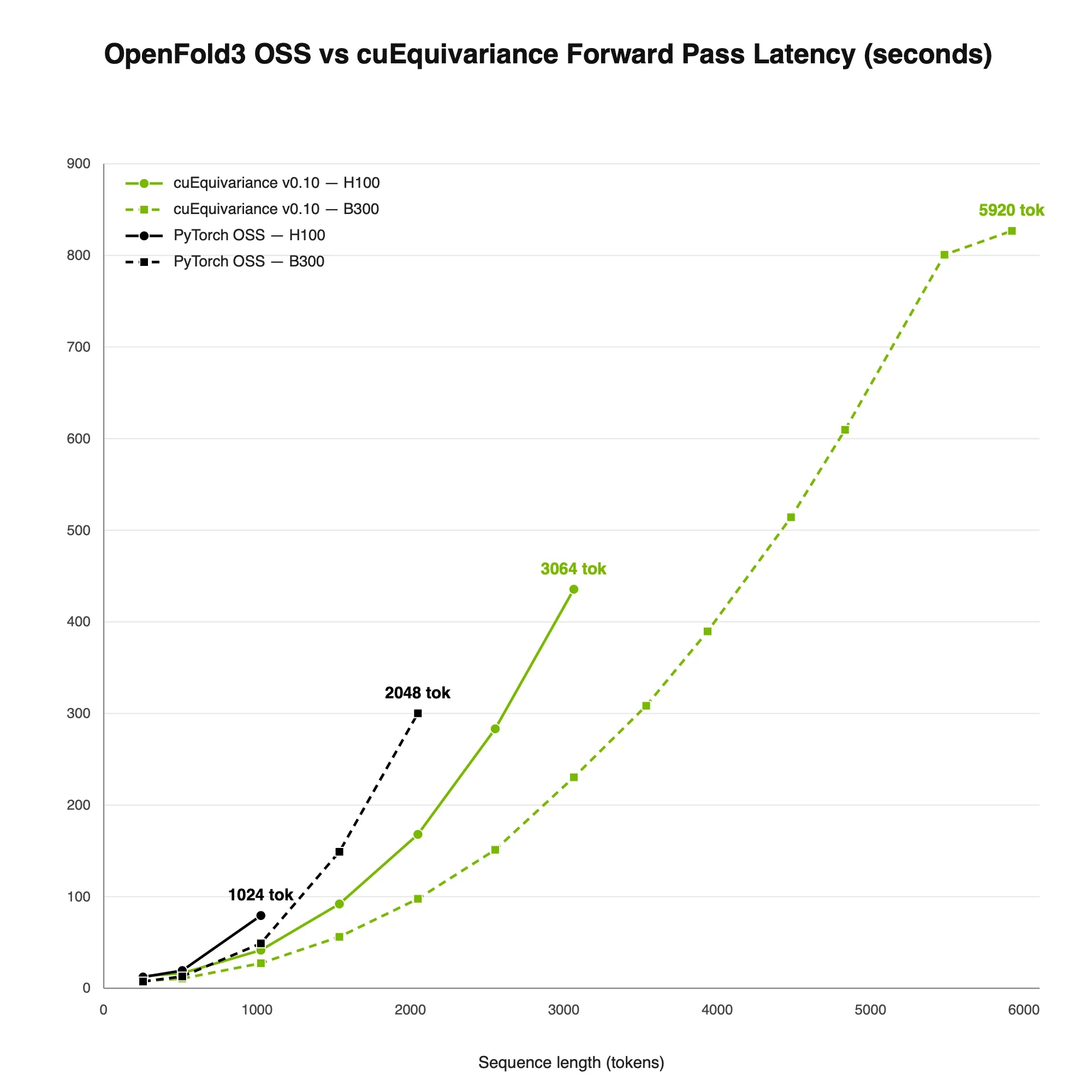 图 3. 在 H100 和 B300 上,相较于 PyTorch,cuEquivariance 可降低延迟并扩展最大序列长度
图 3. 在 H100 和 B300 上,相较于 PyTorch,cuEquivariance 可降低延迟并扩展最大序列长度除了 cuEquivariance 之外,OpenFold3 NIM 还应用了进一步的推理优化来复合增益 (见下文图 4),在单个 B300 上可实现多达 6400 个序列长度。这些额外的加速通过 NIM 提供;对于开箱即用的 SOTA,开发者可以直接调用 NIM 端点或将其组合成智能体工作流。
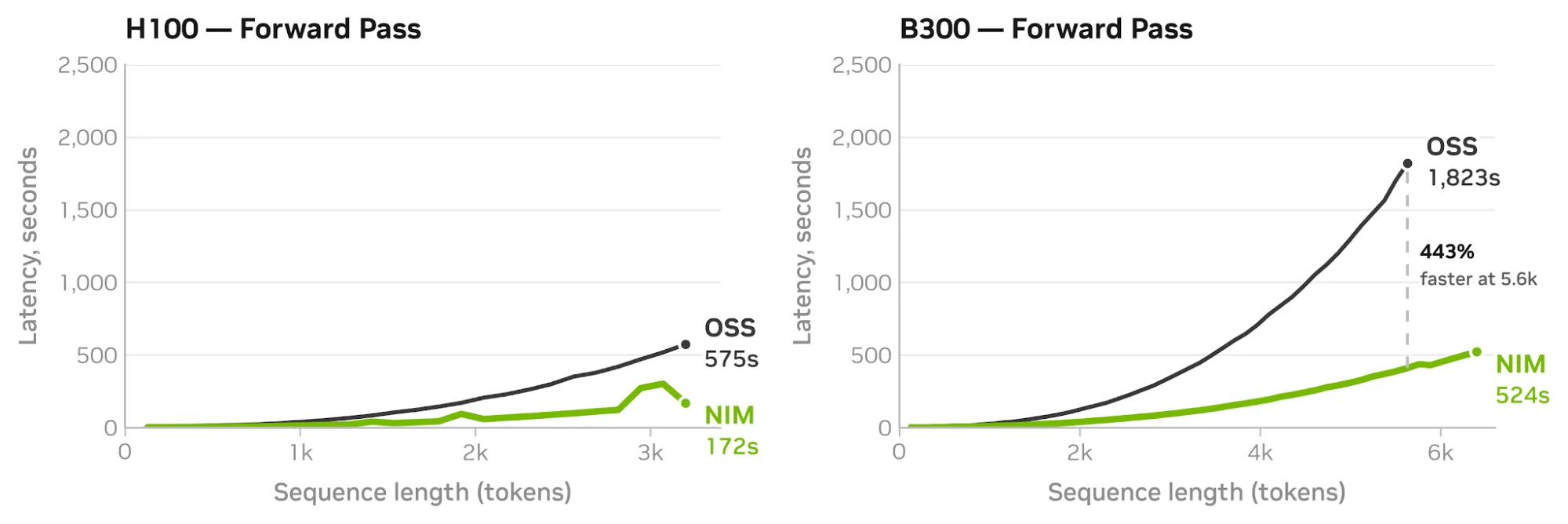 图 4. H100 和 B300 上经过全面优化的 OpenFold3 NIM 分别实现了超过 3 倍和 4 倍的速度提升,在单个 B300 上可折叠多达 6400 个 token
图 4. H100 和 B300 上经过全面优化的 OpenFold3 NIM 分别实现了超过 3 倍和 4 倍的速度提升,在单个 B300 上可折叠多达 6400 个 token # Add the OpenFold3 NIM skill to your agent
npx skills add NVIDIA-BioNeMo/bionemo-agent-toolkit --skill openfold3-nim --agent claude-code
# Hosted API on build.nvidia.com
# You can also use the skill to download the NIM container and provide self-hosted API endpoint. We don't cover that in this tutorial.
export NVIDIA_API_KEY=<key from build.nvidia.com>
# Prompt the agent:
# You have to download a sample target.fasta file. You can prompt the agent to download it for you or point to an already existing file.
"Fold target.fasta with OpenFold3 using the MSA from the previous step; return the ranked structures with confidence scores."
使用 Fold-CP 扩展至多个 GPU
单 GPU 显存在过去一直限制着共同折叠模型的数千个残差。在 NVIDIA B300 (Blackwell Ultra) 上,更大的 HBM 和 Blackwell 生成效率与上述 cuEquivariance 和高级推理优化相结合,在不更改模型的情况下大幅提高了上限。
在许多情况下,单台设备受限可能并不足够。Fold-CP 引入了一种新的并行技术,使每个设备的内存需求扩展为 O(N²/P) 其中 N 是令牌数量,P 是 GPU 数量,在使用玻尔兹 – 2 模型时,在 64 个 B300 上可达到 32000 个令牌,比单 GPU 限制大约跳跃 12×。
要尝试 Fold-CP,只需将您的智能体指向玻尔兹 – CP 代码库,并要求其运行多 GPU 推理。
# Context-parallel inference across 4 GPUs with Fold-CP (boltz-cp)
git clone https://github.com/NVIDIA-Digital-Bio/boltz-cp && cd boltz-cp
# Install dependencies and then run the command below
torchrun --nnodes 1 --nproc_per_node 4 \
src/boltz/distributed/main.py predict /path/to/preprocessed_data \
--out_dir ./predictions \
--size_dp 1 --size_cp 4 \
--recycling_steps 3 --sampling_steps 200 --diffusion_samples 5
加速端到端联合折叠工作流
结构预测性能现已成为端到端系统问题。对于 OpenFold3,实际工作流涵盖 MSA 生成、联合折叠推理、部署,以及决定生物组合建模规模的内存限制。
NVIDIA 可加速该工作流的每一层:MSA Search NIM 可将同源搜索速度提高 177 倍,在 Blackwell GPU 上,cuEquivariance 和 OpenFold3 NIM 可将推理延迟最多降低 4 倍,而 Fold-CP 则展示了上下文并行如何将联合折叠从单个 GPU 扩展到 64 个 NVIDIA B300 GPU 上的 32000 个 token 复合体。
这些工具共同使结构预测更快、更具可扩展性,并更容易组合成代理式发现工作流,帮助研究人员从模型预测转向更大规模、更实用的生物系统。
这些加速解锁了什么
这些改进开辟了以前遥不可及的结构生物学问题类别。在虚拟筛选中,更快的协同折叠推理意味着基于结构的方法 (过去仅用于药物研发活动的最后阶段) 现在可以应用于更早的阶段和更大的库规模,从而提高通过工作流推进的结果的质量和多样性。
对于大型生物分子组件,B300 上扩展的单 GPU 容量与上下文并行的 Fold-CP 框架相结合,改变了可建模的内容:核糖体、剪接体或大型信号组件规模的复合体在结构上难以用于联合折叠模型,而这些加速开始改变这种情况。为了使这种情况具体化,折叠大约 1 万个残基 (大致相当于细菌核糖体的规模) 的复合体的成本会令人望而却步,或者使用单个 GPU 根本无法做到;在使用 Fold-CP 的 B300 上,这种预测可以在多 GPU 节点上实现。
这些是结构生物学在计算方面可能提出的问题的质变,而不仅仅是提高吞吐量。通过开源集成和代理式 API 提供这些工具,将加快计算预测转化为生物学见解并最终转化为新药的速度。
开始使用
试用使用 NVIDIA BioNeMo Agent Toolkit 的加速 OpenFold3 工作流,首先使用以下工具:
- MSA 搜索 NIM: https://build.nvidia.com/colabfold/msa-search
- cuEquivariance:https://github.com/nvidia/cuequivariance
- OpenFold3 NIM:https://build.nvidia.com/openfold/openfold3
- 适用于大型组件的 Fold-CP:https://github.com/NVIDIA-BioNeMo/boltz-cp
致谢
在此,我们要感谢更广泛的 NVIDIA 团队开发基准测试和工具,他们是:Franco Pellegrini、Lalit Vaidya、Duc Tran、Tien Pham、Maximilian Stadler、Alejandro Chacon、Quan Vu、Simon Chu、Brian Roland、Dejun Lin、Joseph Chang、Hoa La、Jonathan Dell、Vishanth Iyer、Timur Rvachov、Christian Dellago、Christian Hundt。我们还要感谢更广泛的 OpenFold 和 OMSF 团队与我们的合作以及他们做出的贡献。